
Infektion und Inflammation bei zystischer Fibrose
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Die Lebenserwartung bei zystischer Fibrose hat sich dank großer therapeutischer Fortschritte gebessert, hochwirksame Kombinationstherapien mit CFTR-Modulatoren können Exazerbationen reduzieren und die Prognose deutlich verbessern. Dennoch bleiben die Keimbesiedlung, insbesondere mit Pseudomonas aeruginosa, sowie neutrophile Entzündungsreaktionen relevante Herausforderungen.
Keypoints
-
Die mediane Überlebensrate bei CF ist auf 66,8 Jahre angestiegen.
-
Bronchopulmonale Exazerbationen sind unter CFTR-Modulator-Therapie signifikant reduziert, treten aber immer noch auf.
-
Menschen mit CF und chronischer Pseudomonas-aeruginosa-Infektion haben weiterhin eine signifikante therapiebedürftige Inflammation und sollten mit inhalativen Antibiotika leitliniengerecht behandelt werden.
-
Studien zu antiinflammatorischen Therapien werden aktuell bereits in klinischen Phasen untersucht.
Einleitung
Die Mukoviszidose/zystische Fibrose (CF) ist eine angeborene Multiorgankrankheit, die autosomal-rezessiv vererbt wird und dereine Dysfunktion oder das Fehlen des epithelialen Ionenkanals CFTR („cystic fibrosis transmembrane conductance regulator“) zugrunde liegt. Die Mutation des Gens, das für CFTR codiert, befindet sich auf dem langen Arm des Chromosoms 7. Durch den Defekt des cAMP(zyklisches Adenosinmonophosphat)-regulierten Kanals ist der Transport von Chloridionen und Bikarbonat an der Zellmembran von Epithelzellen der Atemwege, im Verdauungstrakt und in anderen Organen verringert.1 Die Krankheit ist bis heute nicht heilbar und geht mit einer deutlich eingeschränkten Lebenserwartung einher, das mediane prognostizierte Überlebensalter liegt bei 66,8 Jahren (Abb. 1).2
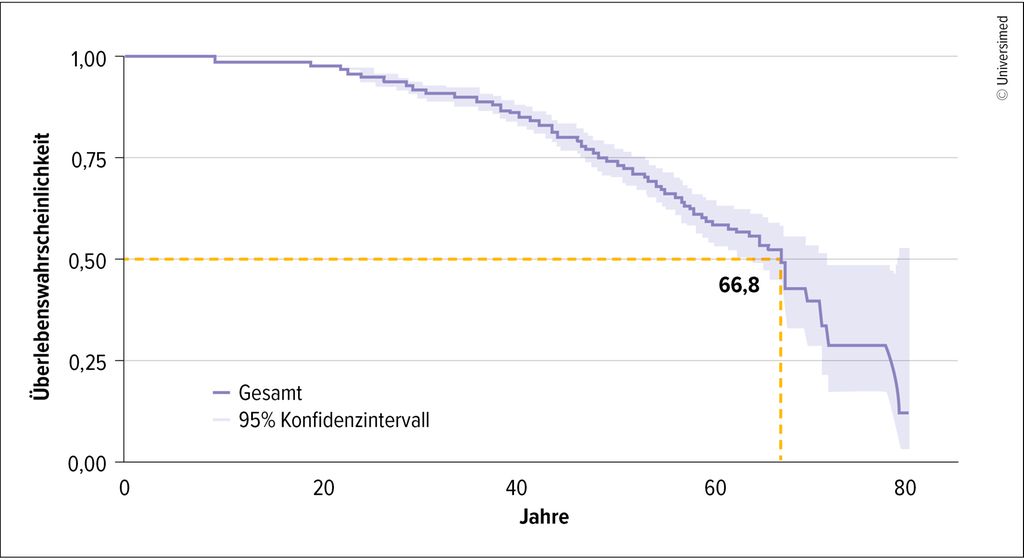
Abb. 1: Medianes Überlebensalter für Mukoviszidose-Patienten im Zeitraum 2020–2024 (Quelle: Deutsches Mukoviszidose-Register 2024)6
Aufgrund einer weitgehend guten klinischen Kontrolle der Erkrankung unter der klinisch sehr effizienten Dreifach-CFTR-Modulator-Therapie3,4 ergeben sich mittlerweile einige Schnittflächen mit der primären Ziliendyskinäsie (PCD), der Bronchiektasenkrankheit, COPD/Asthma und den Immundefektkrankheiten. Hierbei ist neben den bekannten bronchopulmonalen Infektionen, die bei all den genannten Krankheiten auftreten können, vor allem die Inflammation ein diagnostischer und therapeutischer Bereich, der sich als interessanter und klinisch relevanter gemeinsamer Ansatz in der Zukunft verstärkt etablieren könnte.
Pulmonale Infektionen, bronchopulmonale Exazerbationen
Die prognostisch relevanten bronchopulmonalen Exazerbationen sind in Deutschland mit der Zulassung der Triple-CFTR-Modulator-Therapie signifikant gesunken. Dies führte bei den damit therapierten Menschen mit zystischer Fibrose (MmCF) zu einer deutlichen Stabilisierung des Krankheitsverlaufs inklusive einer Verbesserung der FEV1 als wichtiger Prognoseparameter.5 Trotz der deutlich gebesserten Prognose und Stabilisierung der Erkrankung ist in den deutschen Registerdaten des Mukoviszidose e.V. ein Abfall der Lungenfunktion (FEV1 vom Soll) zu erkennen und auch eine signifikante Besiedlung mit Pseudomonas aeruginosa und Staphylococcus aureus (Abb.2 und 3).6 Aus diesem Grund sollten pulmonale Infekte respektive bronchopulmonale Exazerbationen frühzeitig erkannt und therapiert werden, um eventuellen Lungengewebezerstörungen und dem beschriebenen Abfall der Lungenfunktion entgegenzuwirken.
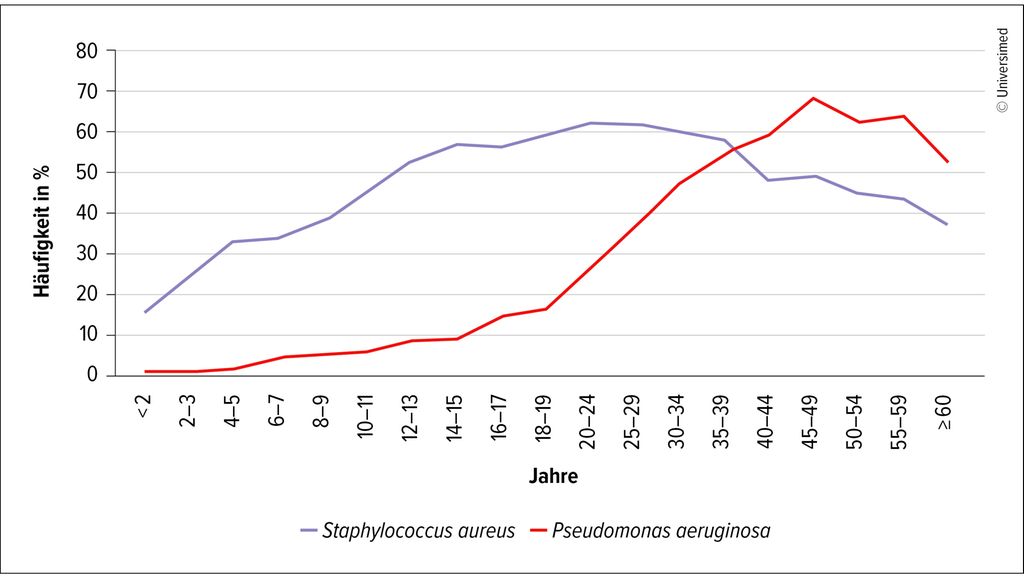
Abb. 2: Chronische Lungeninfektionen bei Mukoviszidose-Patienten mit mikrobiologischer Untersuchung 2024 (Quelle: Deutsches Mukoviszidose-Register)6
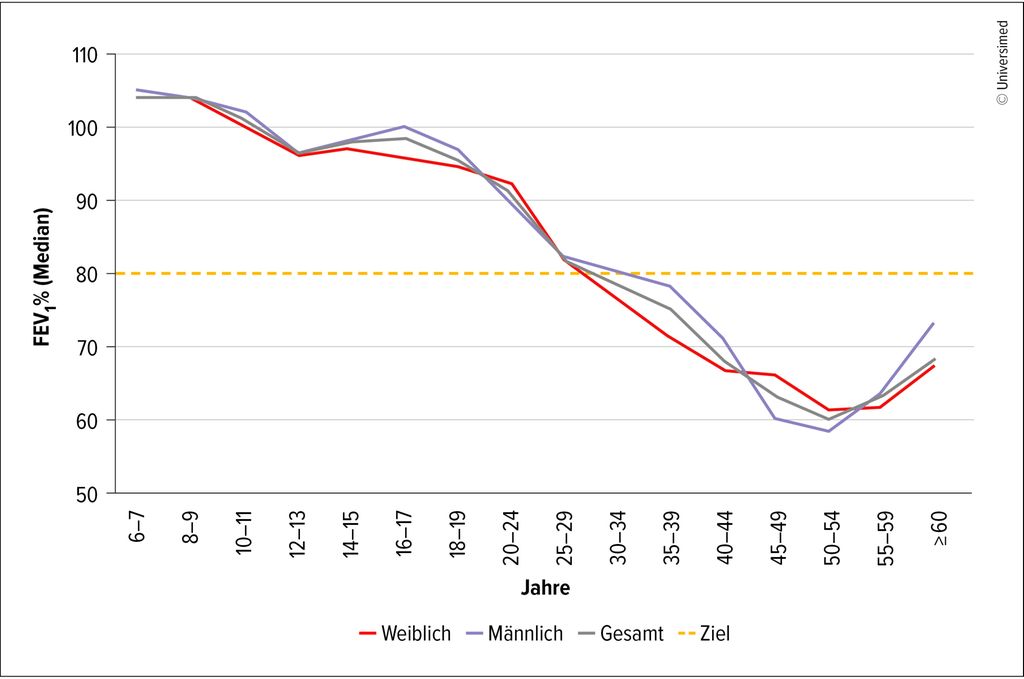
Abb. 3: FEV1%-Wert 2024 (Quelle: Deutsches Mukoviszidose-Register)6
Für chronisch besiedelte MmCF gilt dementsprechend nach der S3-Leitlinie „Lungenerkrankung bei Mukoviszidose: Pseudomonas aeruginosa“auch weiterhin die Empfehlung einer Suppressionstherapie mit einem der vier zugelassenen inhalativen Antibiotika (Tobramycin, Levofloxacin, Colistin, Aztreonam).7 Zusätzlich bestehen neben der inhalativen Antibiotikatherapie weiterhin klare Indikationen für eine orale und auch für eine intravenöse Antibiotikatherapie bei bronchopulmonalen Infektionen, wobei die Art der Antibiotikazuführung vom klinischen Zustand des Patienten abhängig ist.7 Im Rahmen der Behandlung von bronchopulmonalen Infektionen ergeben sich aber neue Fragen, die aktuell noch nicht beantwortet werden können. Hierzu zählt sicherlich die Dosierung der Antibiotika bei einer älter werdenden CF-Population, aber auch die Dauer der Therapie. Aus diesem Grund wären klinische Studien zur oralen und intravenösen Antibiotikatherapie bei MmCF sehr wichtig, die eventuelle Komorbiditäten wie Niereninsuffizienz, kardiovaskuläre Erkrankungen, Lebererkrankungen und zusätzlich das Alter berücksichtigen.
Die Rolle der Inflammation
Trotz der zuvor genannten hochwirksamen Dreifach-CFTR-Modulator-Therapie persistiert bei einem Teil der MmCF eine chronische mikrobielle Besiedlung der Atemwege als wesentlicher Treiber der neutrophilen Entzündungsreaktion. Registeranalysen zeigen zwar eine deutliche Reduktion der Nachweisraten typischer CF-Pathogene unter Elexacaftor/Tezacaftor/Ivacaftor, jedoch bleiben chronische Atemwegsinfektionen bei einem relevanten Anteil der Betroffenen weiterhin bestehen.8
Die zugrunde liegende Entzündungsreaktion ist durch erhöhte Konzentrationen neutrophiler Proteasen sowie proinflammatorischer Mediatoren wie IL-1β, IL-8 und IL-18 gekennzeichnet. Zudem wurden eine verstärkte Aktivierung von Inflammasom-Signalwegen sowie Stressreaktionen des endoplasmatischen Retikulums infolge fehlgefalteter CFTR-Proteine beschrieben.9 Insbesondere die chronische Kolonisation durch gramnegative Keime wie Pseudomonas aeruginosa, Burkholderia cepacia oder Stenotrophomonas maltophilia ist mit einer beschleunigten Lungenfunktionsabnahme und einer erhöhten Exazerbationsrate assoziiert und erfordert bei dauerhaftem Nachweis in der Regel eine langfristige suppressive inhalative Antibiotikatherapie.7,10 Erfreulicherweise zeigt sich nach Beginn einer CFTR-Modulator-Therapie ein Abfall der Inflammation. Allerdings ergibt eine Aufschlüsselung nach Besiedlung mit Pseudomonas aeruginosa bzw. ohne Besiedlung eine persistierende inflammatorische Immunantwort mit wichtigen Inflammationsmarkern wie erhöhtem IL-1β, IL-8, neutrophiler Elastase oder IL-17A bei Pseudomonas-aeruginosa-positiven MmCF.11 In diesem Kontext können die aktuell laufenden antiinflammatorischen Therapien wie zum Beispiel mit Dipeptidyl-Peptidase-1(DPP-1)/Cathepsin-C-Inhibitoren bei CF eine wichtige Rolle spielen.12
Neben den bakteriellen Infektionen spielen bei MmCF auch fungale Infektionen und die allergisch-bronchopulmonale Aspergillose (ABPA) eine wichtige klinische Rolle als Differenzialdiagnosen im Kontext der Infektionen und der Inflammation bei CF.13 Unterschieden werden können bei dem klinisch häufigsten Fadenpilz bei MmCF, dem Aspergillus fumigatus (Af), die Bronchitis, die Pneumonie, die Kolonisation, die Sensibilisierung, die ABPA und das seltene Aspergillom.14 Hierbei spielt die ABPA sicherlich die größte Rolle im klinischen Setting, da diese sehr häufig auftritt, schwere Symptome auslösen kann und lange Therapien nach sich zieht.15 Interessanterweise konnte bei der ABPA in immunologisch-klinischen Studien eine erhöhte Aspergillus-fumigatus-spezifische Immunantwort gezeigt werden. Insbesondere wurde nachgewiesen, dass neben einer bekannten allergischen TH2-Immunantwort auch eine inflammatorische Immunantwort mit erhöhten Aspergillus-fumigatus-spezifischen IL-17a-Werten bei Patienten mit CF und ABPA auftreten kann.16 Mit diesen Forschungsergebnissen konnte erstmals neben der rein allergischen Komponente der ABPA auch eine inflammatorische Immunreaktion bewiesen werden.
Neue diagnostische und therapeutische Möglichkeiten
Dies ermöglicht es, auch hier neue diagnostische Möglichkeiten sowie neue, innovative antiinflammatorische Therapien zum Beispiel mit monoklonalen Antikörpern zu entwickeln.
Die Strategie, einzelne Inflammationsmarker als Biomarker zu identifizieren und anschließend eine zielgerichtete Therapie („targeted therapy“) auf individueller Basis zu etablieren, wird bereits für viele Krankheiten erfolgreich eingesetzt und könnte die Basis für einen Transfer auch zur zystischen Fibrose darstellen. In diesem Kontext könnten folgende Biomarker und Biologika von klinischer Relevanz sein: IL-5 (Reslizumab, Benralizumab, Mepolizumab), IL-4 (Dupilumab), IL-13 (Lebrikizumab, Tralokinumab), IL-8/CXR2 (Navarixin, Danirixin), IL-6 (Tocilizumab), IL-1β (Canakinumab, Anakinra), IL-17/IL-23 (Risankizumab).17
Mit hoher Wahrscheinlichkeit werden sich antiinflammtorische Therapien bei der zystischen Fibrose in der Zukunft klinisch etablieren und eine nützliche Ergänzung zur aktuellen Standardtherapie darstellen.
Zusammenfassung
CF ist eine unheilbare, angeborene Multiorganerkrankung. Es zeigen sich zunehmend Überschneidungen mit anderen Lungenerkrankungen, besonders im Bereich der Inflammation. Aber dank der Dreifach-CFTR-Modulator-Therapien hat sich die Lebenserwartung deutlich verbessert und schwere bronchopulmonale Infektionen und Exazerbationen sind seltener geworden. Dennoch müssen Infektionen früh erkannt und behandelt werden, um Lungenschäden zu vermeiden. Bei chronischer Besiedelung mit Pseudomonas aeruginosa wird weiterhin eine suppressive Therapie mit inhalativen Antibiotika empfohlen. Neue offene Fragen betreffen die Dosierung und die Therapiedauer bei der älter werdenden CF-Population.
Trotz verbesserter Therapien bleibt bei einigen Patienten eine chronische Keimbesiedlung bestehen, die eine starke neutrophile Entzündungsreaktion auslöst. Diese ist gekennzeichnet durch erhöhte Entzündungsbotenstoffe (z.B. IL-1β, IL-8). Besonders die Besiedlung mit Pseudomonas aeru-ginosa ist mit einer anhaltenden Entzündung verbunden. Daher könnten neue antiinflammatorische Wirkstoffe (z.B. DPP-1-Inhibitoren) zukünftig eine wichtige Rolle spielen.
Pilzinfektionen wie z.B. mit Aspergillus fumigatus sind ebenfalls klinisch relevant, insbesondere die allergisch-bronchopulmonale Aspergillose (ABPA). Aktuelle Forschungsergebnisse zeigen, dass bei der ABPA neben einer allergischen auch eine entzündliche Immunreaktion (z.B. über IL-17A) vorliegt – was neue diagnostische und therapeutische Ansätze (z.B. mit Antikörpern) eröffnet.
Ausblick
Die gezielte Hemmung einzelner Entzündungsmarker („targeted therapy“) mit Biologika (z. B. gegen IL-5, IL-4, IL-1β oder IL-17) könnte in Zukunft auf individueller Basis eine sinnvolle Ergänzung der Standardtherapie bei CF darstellen.
Literatur:
1 Ratjen F et al.: Cystic fibrosis. Nat Rev Dis Primers 2015; 1:15010 2 Mukoviszidose e.V.: Neue Daten aus dem Deutschen Mukoviszidose-Register veröffentlicht: Lebenserwartung steigt auf 67 Jahre. https://www.muko.info/einzelansicht/neue-daten-aus-dem-deutschen-mukoviszidose-register-veroeffentlicht-lebenserwartung-steigt-auf-67-jahre ; zuletzt aufgerufen am 21.5.2026 3 Middleton PG et al.: Elexacaftor-tezacaftor-ivacaftor for cystic fibrosis with a single phe508del allele. N Engl J Med 2019; 381: 1809-19 4 Heijerman HGM et al.: Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet 2019; 394: 1940-8 5 Sutharsan S et al.: Impact of elexacaftor/tezacaftor/ivacaftor on lung function, nutritional status, pulmonary exacerbation frequency and sweat chloride in people with cystic fibrosis: real-world evidence from the German CF Registry. Lancet Reg Health Eur 2023; 32: 100690 6 Nährlich L et al.: Deutsches Mukoviszidose-Register. Berichtsband 2024. https://www.muko.info/fileadmin/user_upload/was_wir_tun/register/berichtsbaende/berichtsband_2024.pdf ; zuletzt aufgerufen am 21.5.2026 7 Schwarz C et al.: CF Lung Disease – a German S3 Guideline: Pseudomonas aeruginosa. Pneumologie 2024; 78(6): 367-99 8 Nichols DP et al.: Pharmacologic improvement of CFTR function rapidly decreases sputum pathogen density, but lung infections generally persist. J Clin Invest 2023; 133(10): e167957 9 Peckham DM et al.: CFTR modulators and inflammation. In: Schwarz C et al. (Hrsg.) Inflammation and infection in cystic fibrosis. Karup: European Cystic Fibrosis Society, 2024. 89-108 10 Nichols DPK et al.: Inflammation and infection in cystic fibrosis. In: Schwarz C et al. (Hrsg.) Inflammation and infection in cystic fibrosis. Karup: European Cystic Fibrosis Society, 2024. 15-28 11 Durfey SL et al.: Pseudomonas infections persisting after CFTR modulators are widespread throughout the lungs and drive lung inflammation. Cell Host Microbe 2025; 33: 1428-45.e4 12 Chalmers JD et al.: Dipeptidyl peptidase 1 inhibition as a potential therapeutic approach in neutrophil-mediated inflammatory disease. Front Immunol 2023; 14: 1239151 13 Schwarz C: Epidemiology of airway infection in cystic fibrosis. In: Schwarz C et al. (Hrsg.) Inflammation and infection in cystic fibrosis. Karup: European Cystic Fibrosis Society, 2024. 125-36 14 Schwarz C et al.: Pulmonary aspergillosis in people with cystic fibrosis. Semin Respir Crit Care Med 2024; 45: 128-40 15 Agarwal R et al.: Revised ISHAM-ABPA working group clinical practice guidelines for diagnosing, classifying and treating allergic bronchopulmonary aspergillosis/mycoses. Eur Respir J 2024; 63(4): 2400061 16 Schwarz C et al.: Antigen specificity and cross-reactivity drive functionally diverse anti-aspergillus fumigatus T cell responses in cystic fibrosis. J Clin Invest 2023; 133(5): e161593 17 Giddings O, Esther CR Jr.: Mapping targetable inflammation and outcomes with cystic fibrosis biomarkers. Pediatr Pulmonol 2017; 52(48): 21-8
Das könnte Sie auch interessieren:

Erklärungsversuch des Etagenwechsels von der Rhinitis zum allergischen Asthma
Der „Etagenwechsel“ von der allergischen Rhinitis zum Asthma ist ein zentrales Konzept in der Allergologie – und zugleich komplexer, als der Begriff vermuten lässt. Nach wie vor sind ...
