
VEXAS-Syndrom
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Nachweis von dysfunktionalen Monozyten und gestörtem Inflammasom weist auf neue Therapiemöglichkeiten.
Über die seltene autoinflammatorische Erkrankung VEXAS-Syndrom ist noch wenig bekannt. Forscher aus Paris haben gezeigt, dass sich die Zahl bestimmter Monozyten-Cluster verringert, dass andere zunehmen und dysfunktional werden und es in der Folge zu Entzündungen und Zelltod kommt. Medikamente, die das Inflammasom und den inflammatorischen Zelltod kontrollieren, könnten zukünftig eine Rolle spielen.
2020 beschrieb eine Arbeitsgruppe um den US-amerikanischen Internisten und klinischen Genetiker David Beck von der New York University das erste Mal das VEXAS-Syndrom als eine autoinflammatorische Erkrankung.2 Das Akronym steht für: Vakuolen (in Vorläuferzellen im Knochenmark), (Ubiquitin-aktivierendes) E1-Enzym, X-chromosomal vererbt, autoinflammatorisch und somatisch. Die Prävalenz wurde anhand eines großen Kollektivs auf 1:14000 geschätzt.3 Zu Beginn wurde das VEXAS-Syndrom nur bei Männern beschrieben, bei denen sich somatische Mutationen im UBA1-Gen nachweisen ließen. Das Gen befindet sich auf dem X-Chromosom und kodiert für UBA1, das „ubiquitin-like modifier activating enzyme“. Dieses Enzym spielt eine entscheidende Rolle bei der Regulierung der Protein-Homöostase und bei diversen zellulären Prozessen. Die französischen Forscher und andere Wissenschaftler fanden das VEXAS-Syndrom aber auch bei Frauen, allerdings war eine X-chromosomale Monosomie ursächlich. Das Syndrom tritt im fortgeschrittenen Erwachsenenalter auf. Es kann sich durch verschiedene, uncharakteristische Symptome äußern. Das sind zum einen solche, die typisch für rheumatische Erkrankungen sind (wie Fieber, Arthralgien, Chondritis und Vaskulitis), zum anderen sind es typische Symptome für hämatologische Krankheiten und myelodysplastisches Syndrom (MDS), darunter makrozytäre Anämie, Thrombozytopenie und progressives Knochenmarkversagen. Zudem treten bei vielen Patienten unterschiedliche dermatologische Veränderungen, etwa ein Sweet-Syndrom, auf. Therapeutisch sind in der Regel hohe Glukokortikoiddosen notwendig. Zu immunsuppressiven Therapien gibt es Fallberichte. Einige Patienten profitieren von Azacitidin, das auch gegen MDS eingesetzt wird, oder von Ruxolitinib, einem Inhibitor der Januskinasen 1 und 2. Die einzige kurative Behandlung ist zurzeit eine allogene Stammzelltransplantation.
Die Hypothese der französischen Arbeitsgruppe war, dass die Mutation des UBA1-Gens eine exzessive inflammatorische Antwort durch vermehrten Zelltod verursacht. Sie prüften ihre Hypothese in einer Kohorte von 40 VEXAS-Patienten mit UBA1-Mutation. Als Vergleich dienten 24 Patienten mit schwerer autoinflammatorischer Erkrankung ohne UBA1-Mutation, 4Patienten mit MDS und 12 gesunde Patienten. Den Forschern gelang es, detailliert die biochemischen Veränderungen nachzuweisen. Unter anderem haben VEXAS-Patienten offenbar eine andere Zusammensetzung von Monozyten-Clustern und eine verringerte Anzahl bestimmter Monozyten im Vergleich zu gesunden Patienten (Abb. 1).
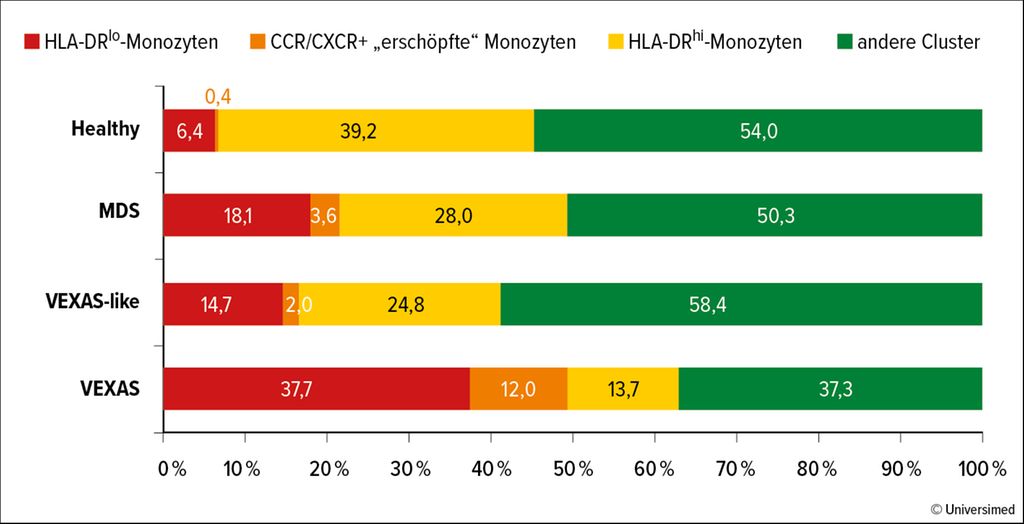
Abb. 1: VEXAS-Patienten zeigen eine andere Zusammensetzung von Monozyten-Clustern im Vergleich zu Gesunden, Patienten mit myelodysplastischem Syndrom (MDS) oder Patienten mit schwerer autoinflammatorischer Erkrankung ohne UBA1-Mutation (VEXAS-like) (nach Kosmider et al. 2024)1
Die Autoren zeigen, dass der Rückgang von Monozyten bei Patienten mit VEXAS-Syndrom mit einer erhöhten Zelltodrate und/oder einer verstärkten Migration in entzündetes Gewebe verbunden sein könnte. Im peripheren Blut bestätigte die RNA-Sequenzierung viele Immunfehlfunktionen und zeigte verstärkte TNF-α- und NFκB-Signalwege. Es ließen sich auch vermehrte Apoptose, Pyroptose und Nekroptose nachweisen. Weitere Studien, die die Analyse von Neutrophilen aus frischen Blutproben ermöglichen, werden nützlich sein, um wichtige Informationen zur Pathophysiologie dieser Störung hinzuzufügen. Die Autoren stellen die Hypothese auf, dass UBA1-mutierte inflammatorische Monozyten mit pathologischerweise exprimierten Zytokinrezeptoren in die Zielgewebe gelockt werden und dort eine lokale Entzündung fördern. Würde man beim Zelltod und in den Signalwegen im Inflammasom gezielt therapeutisch intervenieren, könnte das eine neue Therapiemöglichkeit für VEXAS-Patienten sein.
Literatur:
1 Kosmider O et al.: Nat Commun 2024; 15: 910 2 Beck DB et al.: N Engl J Med 2020; 383: 2628-38 3 Beck DB et al.: JAMA 2023; 329(4): 318-24
Das könnte Sie auch interessieren:
Atypische Frakturen & Bisphosphonate
Bisphosphonate sind Standard in der Osteoporosetherapie und wirksam in der Prävention osteoporotischer Frakturen. Allerdings sind sie in seltenen Fällen mit atypischen Frakturen, vor ...

Selten, aber relevant: Methotrexat kann Knochen schädigen
Bei Schmerzen und Insuffizienzfrakturen der unteren Extremität unter Therapie mit Methotrexat besteht der Verdacht auf eine seltene, aber relevante MTX-Osteopathie. Rheumatolog:innen ...

Osteologische Mythen im Faktencheck
Rund um die Osteoporose und ihre Behandlung kursieren nicht nur unter Betroffenen, sondern selbst in Fachkreisen einige falsche Annahmen. Diese halten sich hartnäckig, obwohl solide ...