Pathobiologie und Genetik der pulmonalen Hypertonie
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Für die 7. Weltkonferenz für pulmonale Hypertonie (World Symposium on Pulmonary Hypertension; WSPH) 2024 beschäftigten sich zwei Task-Forces aus 17 internationalen Experten allein mit den pathologischen Mechanismen, aufbauend auf den biologischen (Task-Force Pathobiology) und den genetischen Grundlagen (Task-Force Genetics) der pulmonalen Hypertonie. Auch wenn die Erkenntnisse und Entwicklungen der letzten Jahre beeindruckend sind, bleiben große Herausforderungen bestehen.
Keypoints
-
Heterogene Pathobiologie erfordert personalisierte Ansätze: Pulmonale Hypertonie (PH) ist eine komplexe Erkrankung mit vielfältigen Ursachen, die von den seltenen idiopathischen Formen bis hin zu den häufigen assoziierten Krankheiten wie Linksherzerkrankungen oder Lungenkrankheiten reichen. Die pathogenetische Vielfalt der PH-Subtypen erschwert die standardisierte Therapie und betont die Bedeutung personalisierter Behandlungsstrategien.
-
Innovative Forschungstechnologien und Biomarker: Fortschritte wie Einzelzell- und räumliche Transkriptomik, Multi-Omics-Ansätze sowie die Entwicklung neuer Biomarker verbessern die Diagnostik und Prognose. Künstliche Intelligenz beschleunigt die Analyse komplexer Datensätze und ebnet den Weg für präzisere Therapieansätze.
-
Genetische und epigenetische Fortschritte: Genetische Mutationen (z.B. in BMPR2, SOX17, TBX4, EIF2AK4) spielen eine Schlüsselrolle in der PH-Pathogenese und eröffnen neue diagnostische und therapeutische Möglichkeiten. Epigenetische Modifikatoren wie Histon-Deacetylasen sind vielversprechende neue therapeutische Ziele, während die genetische Beratung zum Standard bei den seltenen PH Formen gehört.
Pathobiologie
Pulmonale Hypertonie (PH) ist eine komplexe und heterogene Erkrankung, die durch pathologische Umbauprozesse in der Struktur der Lungengefäße gekennzeichnet ist. Insbesondere kleine Arterien, aber auch kleine Venen sind betroffen, was zu einem Anstieg des pulmonalen Gefäßwiderstands (PVR) führt. Dies belastet den rechten Ventrikel und kann letztlich zur Rechtsherzdekompensation führen.
Die Ursachen der PH sind vielfältig und noch nicht vollständig verstanden. Wir sprechen daher weiterhin von idiopathischen Formen ebenso wie von Krankheitszuständen, bei denen die PH mit anderen Erkrankungen assoziiert ist (Tab. 1), beispielsweise chronischer Herzinsuffizienz, chronischen Lungenkrankheiten oder Bindegewebskrankheiten.1

Tab. 1: Klinische Klassifikation der pulmonalen Hypertonie (modifiziert nach Kovacs G et al. 2024)1
Pathologische Veränderungen und histologische Vielfalt
Ein zentraler Mechanismus der PH ist das vaskuläre Remodeling. Charakteristisch sind plexiforme Läsionen, die insbesondere bei der idiopathischen pulmonal- arteriellen Hypertonie (IPAH) auftreten. Gleichzeitig finden sich stets Zellvermehrungen in den Gefäßwänden. Die biologische Vielfalt zwischen den PH-Subtypen erschwert die Entwicklung standardisierter Therapien. So dominieren bei IPAH meistens obliterative Läsionen mit massiver Intimafibrose, während Patienten mit chronisch obstruktiver Lungenkrankheit (COPD) oder idiopathischer pulmonaler Fibrose (IPF) vermehrt eine Gefäßmuskularisierung zeigen. Allerdings gibt es große individuelle Unterschiede. Diese Diversität erklärt die Notwendigkeit spezifischer Behandlungsansätze.
Molekulare Mechanismen und epigenetische Einflüsse
Im Zentrum der pathologischen Mechanismen steht die Dysregulation einiger molekularer Signalwege. Gegenwärtig ist der Fokus auf den „Transforming growth factor (TGF)-β/bone morphogenetic protein“(BMP)-Signalweg gerichet. Genetische Mutationen, etwa im BMP-Rezeptor-2- (BMPR2)-Gen, und eine überaktive Aktivin-Signalgebung fördern sowohl das vaskuläre Remodeling als auch entzündliche Prozesse. Aufbauend auf präklinischen Studien mit Aktivin-Inhibitoren2 wurde kürzlich ein Aktivin-Inhibitor zur Behandlung der PAH zugelassen3,der bei schwerer PAH stärkere klinische Effekte zeigt, als das vorhergesagt wurde.
Epigenetische Veränderungen spielen ebenfalls eine wichtige Rolle. Erhöhte DNA-Methylierung und Histon-Acetylierung fördern pathologische Umbauprozesse. Enzyme wie die Histon-Deacetylase (HDAC) und die Histon-Methyltransferase EZH2 sind bei PH hochreguliert. Inhibitoren dieser epigenetischen Modifikatoren haben in präklinischen Modellen zu positiven Effekten geführt und bieten potenzielle Ansätze für künftige Therapien.
Metabolische Anpassungen und neue Biomarker
Metabolische Veränderungen, etwa eine verstärkte Produktion von Acetyl-CoA, beeinflussen epigenetische Modifikationen wie die Histon-Acetylierung und begünstigen so die Zellproliferation. Weitere molekulare Zielstrukturen wie die RNA-Modifikation N6-Methyladenosin sowie „long non coding (lnc)RNA“ wurden als wichtige Faktoren der PH-Pathogenese identifiziert.
Die Identifikation neuer Biomarker ist von großer Bedeutung für die Verbesserung von Diagnostik und Prognose. Neben etablierten Markern wie dem „brain natriuretic peptide“ (BNP) zeigen zirkulierende Faktoren wie GDF-15, Interleukin-6 und Aktivin A interessante Ergebnisse,4 aber auch metabolische Faktoren wie erhöhte Fettsäuren gewinnen immer mehr an Bedeutung.5
Moderne Technologien wie künstliche Intelligenz (KI) und maschinelles Lernen beschleunigen die Analyse komplexer Datensätze und ebnen den Weg für präzisere, personalisierte Therapien.
Ionenkanäle
Ionenkanäle spielen eine zentrale Rolle bei der Regulierung des pulmonalen Gefäßtonus und der Pathogenese der pulmonalen Hypertonie (PH). Aktuelle Entwicklungen in der Forschung haben spezifische Kalium-, Kalzium- und Natriumkanäle ins Zentrum gerückt. Dysfunktionen in Kaliumkanälen tragen zur Vasokonstriktion und Proliferation von glatten Muskelzellen bei, wodurch der Gefäßwiderstand steigt. Diese Mechanismen sind beispielsweise bei der Wirkung von Prostacyclin und Endothelin beteiligt6 und weitere therapeutische Strategien sind in Entwicklung.7
Ein weiteres Forschungsfeld fokussiert auf „Transient receptor potential“(TRP)-Kanäle, die eine wichtige Rolle bei der intrazellulären Kalziumhomöostase spielen. Überaktive TRP-Kanäle fördern Entzündungen und vaskuläres Remodeling, weswegen TRP-Inhibitoren derzeit in präklinischen Studien untersucht werden.
Geschlechtsspezifische Unterschiede
Ein unzureichend verstandenes Phänomen ist die Tatsache, dass Frauen zwar häufiger von IPAH betroffen sind als Männer, dafür aber länger mit der Krankheit leben können. Das Zusammenwirken der unterschiedlichen genetischen, hormonellen und epigenetischen Faktoren ist sehr komplex. Östrogene spielen eine duale Rolle, da sie einerseits das vaskuläre Remodeling fördern, andererseits aber die Anpassungsfähigkeit des rechten Ventrikels verbessern.
Fortschritte in der Forschung
Technologische Innovationen wie Einzelzell- und räumliche Transkriptomik haben das Verständnis der Pathogenese der PH revolutioniert. Diese Methoden ermöglichen die Identifikation neuer Zellpopulationen und molekularer Marker, wodurch bisher unbekannte Mechanismen aufgedeckt wurden. Multiomics-Ansätze beleuchten die molekulare Heterogenität der PH-Läsionen und liefern neue therapeutische Zielstrukturen.
Modelle und neue Ansätze zur Krankheitsuntersuchung
Die Nutzung induzierter pluripotenter Stammzellen (iPSC) hat zur Entwicklung humaner Krankheitsmodelle geführt, die die Mechanismen der PH in vielen Aspekten besser abbilden können als Tiermodelle. Komplexe 3D-Kulturmodelle wie „lung-on-a-chip“ oder Organoide bieten Möglichkeiten, Zellinteraktionen präzise zu untersuchen. Präzisionsgeschnittene Lungenschnitte (PCLS) erlauben es, In-vivo-Veränderungen der Gefäßstruktur zu simulieren und neue Medikamente zu testen.
Herausforderungen und Ausblick
Trotz der erzielten Fortschritte bleibt die eigentliche Ursache der idiopathischen PAH unbekannt und die Heterogenität der Erkrankung stellt eine große Herausforderung dar. Die Integration individueller Patientendaten in Forschung und Therapieansätze ist essenziell. Eine Verbesserung der präklinischen Modelle und Fortschritte in der Biomarkerforschung könnten den translationalen Erfolg beschleunigen. Technologische Ansätze wie räumliche Transkriptomik und KI-gestützte Analysen könnten personalisierte Therapien weiter voranbringen und die Krankheitslast langfristig reduzieren.
Genetische Entwicklungen und pulmonale Hypertonie
Die genetische Forschung hat seit der Entdeckung der BMPR2-Mutationen vor 25 Jahren eine zunehmende Bedeutung erlangt. Genetische Mutationen sind bei der hereditären pulmonalarteriellen Hypertonie (HPAH) von zentraler Bedeutung, aber häufig zeigt erst die genetische Untersuchung, dass es sich nicht um eine idiopathische, sondern um eine hereditäre PAH handelt. Daher wird für alle Patienten mit HPAH, IPAH und Personen mit kongenitalen Lungen- oder Herzkrankheiten und PH eine genetische Untersuchung empfohlen. Auch bei der seltenen „pulmonary veno-occlusive disease“ (PVOD) kann eine kausale Genmutation vorliegen.8
Obwohl die BMPR2-Mutationen die häufigste Ursache der HPAH darstellen, können Mutationen in vielen weiteren Genen ursächlich sein. Mutationen an SOX17, TBX4, KDR und EIF2AK4 verursachen spezifische PH-Phänotypen. So wurden SOX17-Mutationen sowohl bei Kindern als auch bei Erwachsenen gefunden. Diese Mutationen beeinflussen über regulatorische Elemente die Endothelzellfunktion und vaskuläre Morphogenese, was einen Ansatzpunkt für präzisere therapeutische Ansätze bietet (Abb. 1).9
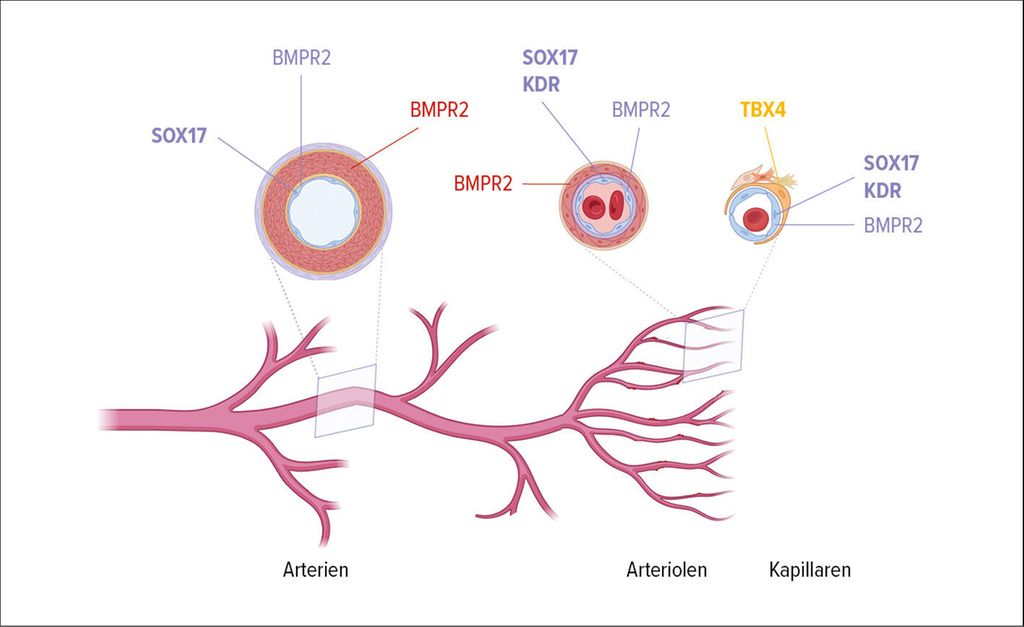
Abb. 1: Neu identifizierte Gene in der Pathobiologie von PAH (erstellt mit https://app.biorender.com)
TBX4-Mutationen sind mit Störungen der Lungenentwicklung assoziiert, darunter reduzierte Alveolarisation und vaskuläres Remodeling. Betroffene zeigen häufig bronchiale Anomalien und parenchymale Veränderungen, die zu einer verminderten Diffusionskapazität (DLCO) führen. Interessanterweise können TBX4-Mutationen sowohl als Verlust- (bei Kindern) als auch als Gewinn-Varianten (bei Erwachsenen) auftreten, was unterschiedliche Krankheitsbilder hervorruft. TBX4-Mutationenstellen nach BMPR2-Mutationen die zweithäufigste Gruppe dar und weisen eine leichte weibliche Prädominanz auf. Diese Mutationen verdeutlichen die Bedeutung von Genen, die die alveolare und vaskuläre Entwicklung steuern, und markieren potenzielle therapeutische Ziele.10
Das KDR-Gen, das für den „vascular endothelial growth factor receptor-2“ (VEGFR-2) codiert, ist ein Schlüsselfaktor der Angiogenese. Mutationen in diesem Gen beeinflussen die Bindung von VEGF-A, einem zentralen Mediator der Gefäßneubildung. KDR-Mutationen wurden sowohl bei Kindern als auch Erwachsenen identifiziert und sind mit vaskulärem Remodeling und schweren pulmonalen Gefäßveränderungen verbunden. Diese Erkenntnisse unterstreichen die Rolle der Angiogenese in der Pathogenese der PH und eröffnen neue therapeutische Möglichkeiten, die gezielt auf VEGFR-2 abzielen könnten.11
Mutationen im EIF2AK4-Gen liegen der hereditären PVOD mit ihrem rezessiven Erbgang zugrunde. Diese Patienten zeichnen sich durch ein niedrigeres Alter bei Diagnose aus, wenn man sie mit den häufigeren Patienten mit idiopathischer PVOD vergleicht.
Zusammen zeigen diese genetischen Mechanismen nicht nur die Vielfalt der genetischen Beiträge zur PH, sondern auch deren potenzielle Bedeutung für die Entwicklung präziserer Diagnose- und Therapieansätze.
Fortschritte in der genetischen Therapie
Die genetische Therapie bietet neue Ansätze für die Behandlung der PH. Methoden wie Gen-Addition, Gen-Editing (z.B. CRISPR-Cas9) und Antisense-Oligonukleotide sind in Entwicklung. Erste Anwendungen dieser Technologien bei anderen Krankheiten, wie z.B. der zystischen Fibrose, bieten Hoffnung auf eine Übertragung auf die PH. Die Herausforderung besteht darin, das geeignete Zeitfenster für therapeutische Interventionen zu definieren, bevor nämlich irreversible Gefäßschäden eingetreten sind.
Klinische Bedeutung und genetisches Testen
Genetische Tests werden zunehmend als Standard bei der Diagnostik der PAH etabliert. Die Identifikation pathogener Varianten hilft nicht nur bei der prognostischen Einschätzung, sondern ermöglicht auch eine fundierte Familienberatung. Genetische Beratung ist essenziell, um reproduktive Entscheidungen und langfristige Überwachungsstrategien zu unterstützen.
Herausforderungen und Ausblick
Obwohl die Fortschritte der letzten Jahre beeindruckend sind, bleiben große Herausforderungen bestehen. Die diverse Pathobiologie und die genetische Heterogenität der PH erfordern größere und diversifizierte Kohorten, um seltene Varianten zu identifizieren und besser zu verstehen. Zukunftsorientierte Ansätze wie die Verwendung der Kombination von KI-gestützten Analysen mit Multiomics könnten bahnbrechende neue Erkenntnisse liefern. Langfristig wird die Präzisionsmedizin nicht nur die Diagnostik und Behandlung, sondern auch die Prävention der PH revolutionieren.
Literatur:
1 Kovacs G et al.: Definition, classification and diagnosis of pulmonary hypertension. Eur Respir J 2024; 64(4): 2401324 2 Guignabert C et al.: Serum and pulmonary expression profiles of the activin signaling system in pulmonary arterial hypertension. Circulation 2023; 147: 1809-22 3 Hoeper MM et al.: (2023) Phase 3 trial of sotatercept for treatment of pulmonary arterial hypertension. N Engl J Med 2023; 388: 1478-90 4 Dardi F et al.: Risk stratification and treatment goals in pulmonary arterial hypertension. Eur Respir J 2024; 64(6): 2401323 5 Bordag N et al.: Lipidomics for diagnosis and prognosis of pulmonary hypertension. medRxiv 2023; https://doi.org/10.1101/2023.05.17.23289772 6 Li Y et al.: Peroxisome proliferator-activated receptor-β/δ, the acute signaling factor in prostacyclin-induced pulmonary vasodilation. Am J Respir Cell Mol Biol 2012; 46: 372-9 7 Csaki R et al.: The TREK-1 potassium channel is a potential pharmacological target for vasorelaxation in pulmonary hypertension. Br J Pharmacol 2024; 181: 3576-93 8 Austin ED et al.: Genetics and precision genomics approaches to pulmonary hypertension. Eur Respir J 2024; 64(4): 2401370 9 Montani D et al.: An emerging phenotype of pulmonary arterial hypertension patients carrying SOX17 variants. Eur Respir J 2022; 60(6): 2200656 10 Karolak JA et al.: Molecular function and contribution of TBX4 in development and disease. Am J Respir Crit Care Med 2023; 207: 855-64 11 Swietlik EM et al.: Bayesian inference associates rare KDR variants with specific phenotypes in pulmonary arterial hypertension. Circ Genom Precis Med 2020; 14(1): e003155
Das könnte Sie auch interessieren:
COPD: neue Leitlinie für eine bessere Patientenversorgung
Die aktualisierte S2k-Leitlinie „Fachärztliche Diagnostik und Therapie der chronisch obstruktiven Lungenerkrankung (COPD) 2026“ wurde im Februar publiziert und beim DGP-Kongress im März ...
