
Im Fokus: VEXAS-Syndrom
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Es tritt vermutlich viel häufiger auf als bisher angenommen: das autoinflammatorische VEXAS-Syndrom. Die chronisch progressiv verlaufende Erkrankung wird zumeist sehr spät diagnostiziert und geht mit einer hohen Mortalität einher. Was ist bislang bekannt zur Pathophysiologie, und welche therapeutischen Ansätze werden derzeit verfolgt?
Keypoints
-
Das VEXAS-Syndrom ist eine komplexe Multisystemerkrankung, die mit Autoinflammation und hämatologischen Veränderungen einhergeht.
-
Zugrunde liegt eine Störung der Ubiquitinierung infolge somatischer Mutationen im UBA1-Gen.
-
Die Therapie des VEXAS-Syndroms ist bislang unbefriedigend und muss sowohl die Autoinflammation als auch die klonale Entartung adressieren.
Beim VEXAS-Syndrom kommt es zu autoinflammatorischen Symptomen sowie Zytopenien. Zudem besteht ein Risiko für die Entwicklung von myelodysplastischen Neoplasmen und multiplem Myelom, erläuterte Prof. Dr. med. Matthias Goebeler, Ärztlicher Direktor der Klinik und Poliklinik für Dermatologie, Venerologie und Allergologie am Uniklinikum Würzburg. Das Akronym VEXAS steht für Vakuolen, E1-Enzym, X-chromosomal, autoinflammatorisch und somatisch. Die Multisystemerkrankung wurde erstmals 2020 beschrieben und entsteht durch eine somatische partielle Loss-of-Function-Mutation im UBA1-Gen, was zu einer variablen Ubiquitinierung und somit einem Ungleichgewicht zwischen Regulatorproteinen führt:1 Ubiquitinierungen sind direkt oder indirekt in zahlreiche zelluläre Prozesse eingebunden, so unter anderem in die Steuerung von Transkription und Translation, Signaltransduktion und DNA-Reparatur, führte Goebeler aus. Dabei wird Ubiquitin auf Zielproteine übertragen, deren Funktionen und Eigenschaften dadurch verändert werden können. Schlüsselenzym bei diesem Prozess ist das «ubiquitin-like modifier activating enzyme 1»(UBA1).
Inflammation durch UBA1-Mutation
Das mRNA-Transkript von UBA1 enthält drei alternative Startcodons an den Positionen M1 (UBA1a), M41 (UBA1b) und M67 (UBA1c). Das Transkript mit M1 enthält das Kernlokalisierungssignal (NLS) und führt zur Translokation von UBA1 in den Zellkern. Die UBA1-mRNA wird auch von Position M41 aus translatiert, wobei das NLS fehlt, sodass die im Zytoplasma verbleibende Isoform UBA1b gebildet wird. Durch Mutationen an Position M41 ist die Translationseffizienz ab M41 reduziert, und es werden mehr Transkripte ab Translationsstart M67 (UBA1c) gebildet, welches aber katalytisch defizient ist.2
Mutationen inUBA1 verringern die Effizienz des endoplasmatischen Retikulum-assoziierten Proteinabbaus (ERAD).3 Dies führt zum Ungleichgewicht in der zellulären Proteostase und zur Anhäufung von ungefaltetem und überflüssigem Protein, das Sensoren der «unfolded protein response» (UPR) aktiviert. Es resultiert eine Aktivierung von Signalkaskaden, die in der Induktion und Aktivierung von Transkriptionsfaktoren (TF) gipfeln. Diese vermitteln die Hochregulierung der Expression von ERAD-Komponenten, von Chaperonen und von Typ-I-Interferonen sowie die Aktivierung des NF-κB-Signalwegs, die wiederum die Autoinflammation vorantreiben.3 Erkrankungen, denen Störungen der Ubiquitinierung zugrunde liegen, rufen also eine systemische Inflammation hervor. Von den inzwischen vierzehn damit assoziierten Krankheitsbildern weisen elf Hauterscheinungen auf, darunter auch das VEXAS-Syndrom, berichtete Goebeler.
Klinische Präsentation
Goebeler präsentierte den Fall eines 54-jährigen Mannes, der sich in der Klinik vorstellte. Der Patient berichtete von einem eruptiven, rumpfbetonten Auftreten von zum Teil sukkulenten Plaques seit einigen Tagen. In den vier vorangegangenen Jahren seien immer wieder selbstlimitierte Schübe gleichartiger Läsionen aufgetreten. Anamnestisch ergaben sich Arthralgien, die zuvor als seronegative rheumatoide Arthritis interpretiert wurden. Eine Therapie mit MTX und Adalimumab blieb ohne Effekt. Der Mann klagte über trockene Augen und gab an, mehrfach mit Antibiotika wegen Chondritis der Ohrknorpel behandelt worden zu sein. Zu guter Letzt berichtete er von rezidivierenden Fieberschüben über 39°C. Die Histologie ergab Sweet-Syndrom-artige Läsionen mit subepidermalem Ödem. Zudem waren deutlich Erythrozytenextravasate und Leukozytoklasie zu erkennen. In einem Biopsat aus einem subkutanen Knoten zeigte sich eine septale Pannikulitis. «Wir hatten kurz zuvor in einem Seminar das erste Mal vom VEXAS-Syndrom gehört und kamen daher auf die entsprechende Verdachtsdiagnose», berichtete Goebeler. In einer Sanger-Sequenzierung des UBA1-Gens konnte die relevante Mutation nachgewiesen werden.
Das VEXAS-Syndrom tritt im fortgeschrittenen Erwachsenenalter vorzugsweise bei Männern auf und ist durch hämatologische, rheumatologische und dermatologische Symptome gekennzeichnet.4 Zu Letzteren gehören neutrophilenreiche Läsionen, die an das Sweet-Syndrom erinnern: Erythema-nodosum- und Pannikulitis-ähnliche Hautmanifestationen sowie rezidivierende Polychondritis der Nase und der Ohrmuscheln. Charakteristisch ist das Vorhandensein von zytoplasmatischen Vakuolen in myeloischen und erythroiden Vorstufen im Knochenmark. «Das Fehlen von Vakuolen schliesst ein VEXAS-Syndrom nicht aus», hob der Experte hervor.
Wie ist die Prognose des VEXAS-Syndroms? Die 5-Jahres-Überlebensrate nach Auftreten der ersten Manifestationen beträgt circa 80 Prozent.5 In bis zu 50 Prozent der Fälle ist die Erkrankung mit einem myelodysplastischen Syndrom (MDS) assoziiert. Bei MDS liegen häufiger zusätzlich Komutationen in den DNMT3A- und TET2-Genen vor, was mit einer schlechteren Prognose assoziiert ist. Zudem lässt sich eine Häufung des multiplen Myeloms beobachten, obwohl Lymphozyten keine UBA1-Mutationen aufweisen.5
Diagnostik und Therapie
Ein klinischer Score zur Abschätzung der Wahrscheinlichkeit unterstützt bei der Diagnosestellung:6 Mit jeweils einem Punkt werden ein Alter >50 Jahre, Hautläsionen, pulmonale Beteiligung und Chondritis bewertet. Eine makrozytäre Anämie zählt zwei Punkte. Ab drei Punkten ist das Vorliegen eines VEXAS-Syndroms möglich, und es sollte eine genetische Abklärung erfolgen (Abb.1).
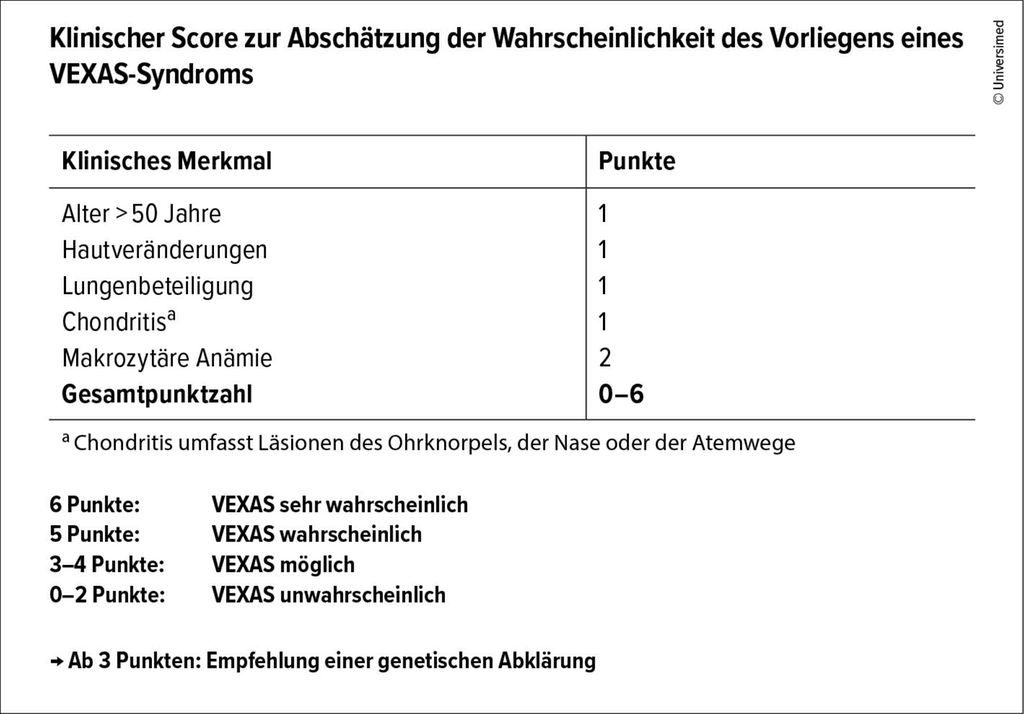
Abb. 1: Ein klinischer Score hilft bei der Diagnosestellung des VEXAS-Syndroms (modifiziert nach Maeda A et al. 2024)6
Die Therapie ist bislang unbefriedigend, meinte Goebeler. Sie müsse sowohl die Autoinflammation als auch die klonale Entartung adressieren. «Am besten funktionieren systemische Glukokortikoide, die jedoch auf lange Sicht zu anderen Problemen führen können», berichtete er. Klassische Immunsuppressiva wie Azathioprin, MMF und MTX wirken laut Goebeler kaum bis gar nicht. Weiters wies er darauf hin, dass bei den Biologika der IL-1-Rezeptor-Antagonist (RA) Anakinra nahezu immer zu ausgeprägten Hautreaktionen führt und nicht eingesetzt werden sollte, TNF-Antagonisten bewirken bestenfalls ein kurzfristiges Ansprechen und der IL-6RA Tocilizumab zeigt nur einen mässigen Effekt. «JAK2-Inhibitoren wie insbesondere Ruxolitinib wirken recht gut auf die Hautläsionen», so der Dermatologe. Bei Zytopenien, Transfusionsbedürftigkeit, MDS und morphologischen Dysplasien lautet die Empfehlung, Azacitidin einzusetzen, das hypomethylierend wirkt. «Der einzige kurative Ansatz wäre eine allogene hämatopoetische Stammzelltransplantation», so Goebeler. Als innovative Therapiestrategien werden derzeit pharmakologische Aktivatoren von UBA1 sowie des Proteasoms diskutiert.3
Quelle:
«Autoinflammatorische Syndrome und das VEXAS-Syndrom: neue Erkenntnisse zu Pathomechanismen und Therapie», Vortrag von Prof. Dr. med. Matthias Goebeler, Würzburg, im Rahmen der DDG-Jahrestagung am 2. Mai 2025 in Berlin
Literatur:
1 Beck DB et al.: Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med 2020; 383(27): 2628-38 2 Sakuma M et al.: UBA1 dysfunction in VEXAS and cancer. Oncotarget 2024; 15: 644-58 3 Beck DB et al.: Disorders of ubiquitylation: unchained inflammation. Nat Rev Rheumatol 2022; 18(8): 435-47 4 Baur V et al.: VEXAS-syndrome, a newly described autoinflammatory systemic disease with dermatologic manifestations. J Dtsch Dermatol Ges 2023; 21(12): 1456-63 5 Koster MJ et al.: VEXAS syndrome: clinical, hematologic features and a practical approach to diagnosis and management. Am J Hematol 2024; 99(2): 284-99 6 Maeda A et al.: Efficient detection of somatic UBA1 variants and clinical scoring system predicting patients with variants in VEXAS syndrome. Rheumatology (Oxford) 2024; 63(8): 2056-64
Das könnte Sie auch interessieren:

Frauenhaut wird anders krank, Männerhaut auch
Asthma wird bei Mädchen später diagnostiziert als bei Jungen, Parkinson und Harnblasenkarzinom bei Frauen später als bei Männern. Auch ein Myokardinfarkt wird bei Frauen immer noch ...
Kombinationstherapie mit plättchenreichem Plasma und Hyaluronsäure
Hochwertiges autologes plättchenreiches Plasma (PRP) verfügt von Natur aus über einen komplex zusammengesetzten Cocktail aus zahlreichen bioaktiven Substanzen. Gegenüber dem Vollblut ...
Mikrobiologische Diagnostik von Wundinfektionen
Die mikrobiologische Diagnostik von Wundinfektionen kann – richtig eingesetzt – ein essenzieller Bestandteil der klinischen Entscheidungsfindung im Rahmen der Behandlung von ...